Standards for Medicines
USP develops and publishes standards for drug substances, drug products, excipients, and dietary supplements in the United States Pharmacopeia–National Formulary (USP–NF). These standards have been recognized in the Federal Food, Drug and Cosmetic (FD&C) Act since it was first enacted in 1938. The FD&C Act defines the term "official compendium” as the official USP, the official NF, the official Homeopathic Pharmacopeia of the United States, or any supplement to them. USP–NF standards play a role in the adulteration and misbranding provisions of the FD&C Act (which apply as well to biologics, a subset of drugs, under the Public Health Service Act). USP has no role in enforcement of these or other provisions that recognize USP–NF standards, which is the responsibility of FDA and other government authorities in the United States and elsewhere. Manufacturers and potentially affected parties are encouraged to contact FDA with questions about the specific applicability of USP standards to their products.
Specific Drug Categories and Topics
- Drugs—USP's goal is to have substance and preparation (product) monographs in USP–NF for all FDA-approved drugs, including biologics, and their ingredients. USP also develops monographs for therapeutic products not approved by FDA, e.g., pre-1938 drugs, dietary supplements, and compounded preparations. Although submission of information needed to develop a monograph by the Council of Experts is voluntary, compliance with a USP–NF monograph, if available, is mandatory in the following respects:
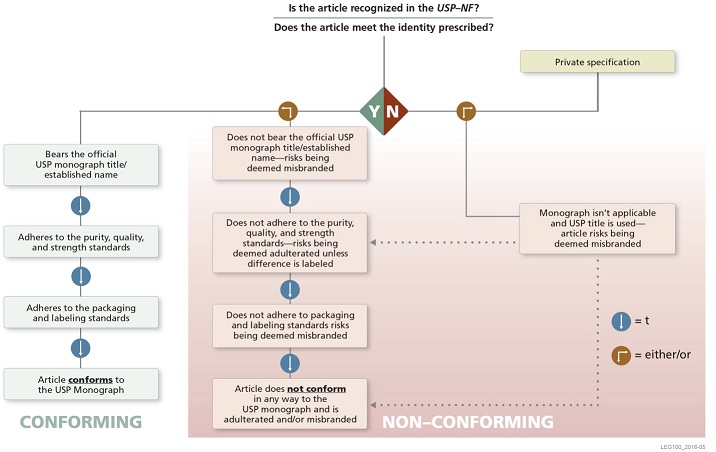
- Nonproprietary Name: Under the relevant FD&C Act provisions, a drug will be deemed misbranded unless its label bears to the exclusion of any other nonproprietary name the "established” name, which ordinarily is the compendial name (see discussion of Nomenclature, below).
- Identity: A drug with a name recognized in USP–NF must comply with the identity/identification requirements of its monograph, or be deemed adulterated, misbranded, or both.
- Strength, Quality, Purity: Drugs also must comply with compendial standards for strength, quality, and purity (tests for assay and impurities), unless labeled to show all respects in which the drugs differ. FDA requires that names for articles that are not official must be clearly distinguishing and differentiating from any name recognized in an official compendium.
- Packaging, Labeling: Drugs with a name recognized in USP–NF also will be considered misbranded unless they meet compendial standards for packaging and labeling.
- Biologics—In the United States, all biologics are considered a subset of drugs, whether they are approved by FDA under the FD&C Act [and receive a new drug application (NDA)] or under the Public Health Service Act [PHS Act, where they receive a biologics license application (BLA)]. As a result, all PHS Act biologics are subject to the drug regulatory requirements of the FD&C Act, which means they are required to comply with the adulteration and misbranding provisions of the FD&C Act, including USP–NF compendial requirements. This is equally so for biologics approved under the longstanding PHS Act "351(a)” pathway, as well as the new "351(k)” pathway for biosimilars added by the 2010 healthcare reform legislation.
The following graphic illustrates the applicability of USP standards to drugs and biologics in the United States.
- Compounded Preparations—Compounding means the preparation, mixing, assembling, altering, packaging, and labeling of a drug, drug-delivery device, or device in accordance with a licensed practitioner's prescription, medication order, or initiative based on the practitioner/patient/pharmacist/compounder relationship in the course of professional practice. USP provides both general chapters and monographs for compounded preparations. Compounded preparation monographs include formulas (ingredients and quantities), specific directions to correctly compound the particular preparation, packaging and storage information, labeling information, pH, beyond-use dates based on stability studies, and detailed assays (majority of monographs). Standards in USP–NF for compounded preparations may be enforced by both the states (as pharmacy practice/compounding is traditionally regulated by state boards of pharmacy), and FDA (as compounded preparations remain subject to the adulteration and misbranding provisions of the FD&C Act which require conformance to certain USP–NF standards).
- Nomenclature—In the United States, FDA generally defers to USP to create established (nonproprietary) names for drug products, including proper names for biologics. USP, as a member of the United States Adopted Names (USAN) Council, works to determine names for drug and biological substances. USP's authority to develop official nonproprietary names is identified in section 502(e) of the FD&C Act. FDA's policy on established names is set forth in 21 CFR 299.4. FDA-approved nonproprietary names are considered by FDA and the courts to be interim names that exist only unless and until USP designates a name. In contrast to USP's role in designating nonproprietary names, the designation of proprietary names and brand names is solely the responsibility of FDA, working with applicants.
The USP Nomenclature Expert Committee was formed in 1986 to create appropriate established names for dosage forms and combination drug products, and to develop naming policies. The Nomenclature Expert Committee coordinates its work with the USAN Council, and it establishes the Pronunciation Guide, which is utilized by USAN.
The USAN Council began in 1961 by providing ingredient names for drugs prior to their marketing. USP participates in this activity, together with the American Medical Association, the American Pharmacists Association, and FDA. The Council's output is incorporated, along with other names for drugs (including generic, proprietary, and chemical names and code designations), in the USP Dictionary of USAN and International Drug Names (the Dictionary). Since 1988, this publication has been recognized by federal regulation as the source of established names for drug substances in the United States (except in those cases where USP specifies a different non-proprietary name).

Standards for Medical Devices
Section 201(h) of the FD&C Act defines a device as an instrument, apparatus, similar article, or component thereof recognized in USP–NF. Section 502(e) of the FD&C Act defines the established name of a device in the absence of an FDA designation of the official name as the official title in an official compendium. Despite these statutory provisions, there is no comparable recognition of USP's role in establishing compendial standards for medical devices as exists for drugs and biologics. Under authority granted by the Food and Drug Administration Modernization Act of 1997, the Center for Devices and Radiological Health recognizes national and international standards, including some USP tests and assays, for medical devices.
Standards for Food Ingredients
USP develops and publishes standards for food ingredients in the Food Chemicals Codex (FCC). Currently, more than 200 FDA food additive regulations incorporate FCC monographs by reference. Conformance to an FCC standard is required where FDA has specifically adopted that standard by regulation and where the ingredient is marketed on the basis of that regulation. Even in the absence of regulatory standards, FCC standards are generally accepted by FDA and the food industry as standards for “food grade” substances.
The first edition of the FCC, which was published in 1966 by the Institute of Medicine, was given quasi-legal recognition in July 1966 by means of a letter of endorsement from FDA Commissioner James L. Goddard, which was reprinted in the book. The letter stated that "the FDA will regard the specifications in the Food Chemicals Codex as defining an 'appropriate food grade' within the meaning of Sec. 121.101(b)(3) and Sec. 121.1000(a)(2) of the food additive regulations, subject to the following qualification: this endorsement is not construed to exempt any food chemical appearing in the Food Chemicals Codex from compliance with requirements of Acts of Congress or with regulations and rulings issued by the Food and Drug Administration under authority of such Acts."
Subsequently, various additional specifications from previous FCC editions were also incorporated by reference, in the U.S. Code of Federal Regulations to define specific safe ingredients under Title 21, in various parts of Sections 172, 173, and 184. It is anticipated that FDA will from time to time continue to update its regulatory references to the FCC.
USP’s goal is to have FCC monographs for all substances added to foods in the U.S., including all ingredients that are marketed as food additives and color additives under an FDA regulation following a successful petition of FDA, ingredients that are affirmed to be GRAS, and ingredients that are marketed under approvals issued prior to the 1958 Food Additive Amendments (prior-sanctioned items).
The existence of an FCC monograph for a food ingredient does not provide independent evidence that a particular product may be lawfully marketed in the United States under the FD&C Act and its implementing regulations. It is the ultimate responsibility of food and ingredient manufacturers and distributors to ensure that their products are legally marketed in the United States.
Standards for Dietary Supplements
USP's standards for dietary supplements and ingredients used in the production of dietary supplements may be found in the USP–NF and the FCC.
- USP–NF Dietary Supplement Standards—The Dietary Supplement Health and Education Act of 1994 amendments to the FD&C Act name the USP and NF as official compendia for dietary supplements. The amendments also provide that a dietary supplement may be deemed misbranded if it is covered by a monograph in an official compendium, is represented as conforming to this monograph, but fails to so conform. The dietary supplement must be represented as conforming to a USP–NF dietary supplement monograph in order for the compendial standards to apply. This contrasts with pharmaceutical products, wherein conformance to the monograph is mandatory whether or not the product claims to conform.
- FCC Dietary Supplement Standards—Dietary supplement standards found in the FCC are considered to be food ingredients and are subject to the same recognition parameters—see "Standards for Food Ingredients” above.
The existence of a monograph for a dietary supplement does not provide independent evidence that a particular product may be lawfully marketed in the United States under the FD&C Act and its implementing regulations. It is the ultimate responsibility of dietary supplement manufacturers and distributors to ensure that their products are legally marketed in the United States.